

Hội chứng Kallmann – gây “không có tinh trùng” ở nam giới
Hội chứng Kallmann là một dạng của thiểu năng sinh dục Hypogonadotropic gây suy giảm khứu giác và dậy thì muộn, xảy ra do thiếu hụt một số hormone định hướng sự phát triển giới tính. Những hormone này thường được tạo ra trong vùng dưới đồi của não.


Đặc điểm lâm sàng
Nam giới bị thiểu năng sinh dục Hypogonadotropic thường có dương vật nhỏ bất thường và tinh hoàn không nằm đúng vị trí (tinh hoàn ẩn).
Ở tuổi dậy thì, hầu hết những người mắc bệnh đều không phát triển các đặc điểm sinh dục thứ cấp, chẳng hạn như sự phát triển của lông mặt và giọng nói trầm hơn ở nam giới, bắt đầu kỳ kinh nguyệt mỗi tháng và sự phát triển vú ở nữ giới cùng với sự phát triển chiều cao ở cả hai giới. Nếu không được điều trị, hầu hết những người mắc bệnh, kể cả nam giới và nữ giới đều không thể có con.
Người người mắc hội chứng Kallmann có khứu giác bị suy giảm hoặc hoàn toàn không có khứu giác (mất khứu giác). Đặc điểm này giúp phân biệt hội chứng Kallmann với hầu hết các dạng thiểu năng sinh dục khác. Đôi khi những người mắc hội chứng Kallmann không biết rằng họ không thể phát hiện ra mùi cho đến khi được phát hiện thông qua xét nghiệm.
Hội chứng Kallmann có thể có nhiều dấu hiệu và triệu chứng khác nhau, bao gồm suy giảm chức năng một bên thận, bất thường ở các xương ngón tay hoặc ngón chân, sứt môi có hoặc không có khe hở ở vòm miệng (hở hàm ếch ), cử động mắt bất thường, mất thính giác, và phát triển rang bất thường. Một số người mắc bệnh có một đặc điểm gọi là hiện tượng đồng động hai tay, trong đó các chuyển động của một tay được phản chiếu bởi tay kia. Hiện tượng đồng động này có thể gây khó khăn khi thực hiện các công việc đòi hỏi hai tay phải cử động riêng rẽ, chẳng hạn như chơi nhạc cụ.

Nguyên nhân
Các đột biến trên hơn 20 gen có liên quan đến hội chứng Kallmann. Trong số các nguyên nhân phổ biến nhất của hội chứng này là do đột biến gen ANOS1, CHD7, FGF8, FGFR1, PROK2 hoặc PROKR2. Trong một số trường hợp, những người mắc bệnh có nhiều hơn một đột biến ở các gen này. Ngoài ra, các nhà nghiên cứu đã xác định được các đột biến trên các gen khác có thể góp phần vào sự phát triển các triệu chứng của hội chứng Kallmann.
Các gen liên quan đến hội chứng Kallmann đóng vai trò trong sự phát triển của một số vùng não trước khi sinh. Mặc dù một số chức năng cụ thể của các gen này vẫn chưa rõ ràng, nhưng dường như chúng có liên quan đến sự hình thành và di chuyển của một nhóm tế bào thần kinh khứu giác. Các tế bào thần kinh này được hình thành từ mũi đang phát triển và sau đó cùng nhau di chuyển đến khu vực phía trước não gọi là hành khứu giác , một vị trí quan trọng đối với việc nhận biết mùi.

Nghiên cứu cho thấy các gen liên quan đến hội chứng Kallmann cũng liên quan đến sự di chuyển của các tế bào thần kinh sản xuất ra một loại hormone được gọi là hormone giải phóng gonadotropin (GnRH). Giống như các tế bào thần kinh khứu giác, các tế bào thần kinh sản xuất GnRH di chuyển từ mũi đang phát triển đến phía trước của não. GnRH kiểm soát việc sản xuất một số hormone hướng dẫn sự phát triển giới tính trước khi sinh và trong tuổi dậy thì. Những hormone này rất quan trọng đối với chức năng của buồng trứng ở phụ nữ và tinh hoàn ở nam giới.
Đột biến gen liên quan đến hội chứng Kallmann làm gián đoạn sự di chuyển của các tế bào thần kinh khứu giác và các tế bào thần kinh sản xuất GnRH trong não đang phát triển. Nếu các tế bào thần kinh khứu giác không di chuyển đến hành khứu giác, khứu giác của một người có thể bị giảm hoặc không hoạt động. Các tế bào thần kinh sản xuất GnRH di chuyển sai vị trí làm ngăn cản việc sản xuất các hormone sinh dục và gây ra các triệu chứng của thiểu năng sinh dục. Không rõ cụ thể các đột biến gen ảnh hưởng như thế nào đến các dấu hiệu và triệu chứng xảy ra trong hội chứng Kallmann. Bởi vì các đặc điểm của hội chứng này khác nhau giữa những người mắc bệnh, các yếu tố di truyền và môi trường có thể góp phần gây ra căn bệnh này.
Đột biến trên các gen đã biết chiếm khoảng 30% tổng số các ca mắc hội chứng Kallmann, những trường hợp khác được xem là chưa xác định được nguyên nhân. Các nhà nghiên cứu đang tìm kiếm các đột biến gen khác có thể gây ra hội chứng này.
Chẩn đoán hội chứng Kallmann
Chẩn đoán hội chứng Kallmann dựa trên biểu hiện lâm sàng về sự phát triển ở tuổi dậy thì theo phân loại Tanner. Phân loại Tanner được thiết lập sử dụng trong quá trình khám sức khỏe của các bác sĩ nội tiết và nhi khoa trên toàn thế giới để đánh giá sự trưởng thành của các đặc điểm sinh dục sơ cấp và thứ cấp.
Những bất thường về sức khỏe gây ra bởi hội chứng này thường xuất hiện khi mới sinh, nhưng có thể được chẩn đoán trễ hơn (giai đoạn dậy thì 14-16 tuổi). Việc chẩn đoán bệnh dựa trên thăm khám lâm sàng, thực hiện các xét nghiệm nội tiết, hình ảnh học và xét nghiệm di truyền.
Dấu hiệu lâm sàng: Các triệu chứng và dấu hiệu dậy thì muộn hay mất khứu giác. Đặc điểm này giúp phân biệt hội chứng Kallmann với hầu hết các dạng thiểu năng sinh dục khác, không ảnh hưởng đến khứu giác.
Xét nghiệm sinh hóa: Là một phần quan trọng trong chẩn đoán hội chứng Kallmann. Hormone GnRH không thể đo lường trực tiếp trong cơ thể thay vào đó sẽ xét nghiệm sẽ nồng độ trong máu của các hormone như LH, FSH và các hormone sinh dục như testosterone, estrogen và progesterone. Các hormone này sẽ giảm thấp trong hội chứng Kallmann
Đồng thời, phối hợp với bác sĩ Trung tâm Chẩn đoán hình ảnh chụp cộng hưởng từ (MRI) để khảo sát vùng dưới đồi, tuyến yên và mũi để tìm nguyên nhân gây ra các bất thường về việc giảm tiết hormone và mất chức năng khứu giác.
Cuối cùng các xét nghiệm di truyền có thể được thực hiện để xác định chẩn đoán bệnh.

Xét nghiệm di truyền, tình trạng đột biến gen ảnh hưởng đến sự phát triển của một số tế bào thần kinh trong não là nguyên nhân gây ra hội chứng Kallmann. Có khoảng 25 gen khác nhau liên quan đến hội chứng này, nhưng phổ biến nhất là tình trạng đột biến trên 6 loại gen ANOS1, CHD7, FGF8, FGFR1, PROK2 và PROKR2. Các đột biến gen này chiếm khoảng 50% trường hợp mắc hội chứng Kallmann. Những trường hợp còn lại có thể không rõ nguyên nhân hoặc do các đột biến mới chưa được phát hiện.
Ngoài ra, chụp MRI vùng dưới đồi-tuyến yên để kiểm tra cấu trúc bên trong não. MRI cũng có thể phát hiện sự vắng mặt của các cấu trúc hành khứu giác ở những người mắc hội chứng Kallmann.
Khứu giác có thể được đánh giá bằng các bài kiểm tra như nhận dạng mùi của Đại học Pennsylvania (UPSIT). Thử nghiệm này đánh giá khả năng cảm nhận 40 chất tạo mùi. Xác định một người mất khứu giác hay suy giảm khứu giác dựa trên kết quả kiểm tra, độ tuổi và giới tính của từng người.
3 cách di truyền của hội chứng Kallmann
1. Di truyền theo kiểu liên kết nhiễm sắc thể giới tính X
Nam giới chỉ cần nhận một nhiễm sắc thể X mang gen đột biến từ mẹ là có thể biểu hiện bệnh. Với nữ giới (người có hai nhiễm sắc thể X) thì cần tới hai gen đột biến. Kiểu di truyền này cũng có nghĩa, người cha không thể truyền bệnh cho con trai (do con trai chỉ nhận nhiễm sắc thể Y từ cha). Một ví dụ về hội chứng Kallmann di truyền theo kiểu này là dạng đột biến gen ANOS1.
2. Di truyền trội trên nhiễm sắc thể thường
Chỉ cần 1 trong 2 người cha hoặc mẹ mang gen đột biến là có thể truyền cho con cái và gây ra bệnh. Người con chỉ cần mang một gen đột biến là có thể biểu hiện bệnh. Hội chứng Kallmann do những đột biến gen sau đây sẽ có kiểu di truyền trội trên nhiễm sắc thể thường, bao gồm: FGFR1, PROKR2, PROK2, CHD7, FGF8.
3. Di truyền lặn trên nhiễm sắc thể thường
Cả cha và mẹ (thường là người không có triệu chứng) có thể sinh ra con bị hội chứng Kallmann. Trong trường hợp này, người con phải mang 2 gen gây bệnh nhận từ cha và mẹ. Các gen bị đột biến trong trường hợp này là PROKR và PROK2.
Dạng di truyền diễn giải theo bản đồ di truyền
Khi hội chứng Kallmann do đột biến gen ANOS1 gây ra, tình trạng này có kiểu di truyền lặn liên kết X. Gen ANOS1 nằm trên nhiễm sắc thể X, là một trong hai nhiễm sắc thể giới tính. Nam giới chỉ có một nhiễm sắc thể X, một bản sao bị đột biến của gen trong mỗi tế bào là đủ để gây ra tình trạng này. Nữ giới có hai nhiễm sắc thể X, cả hai bản sao của gen bị đột biến mới đủ để gây ra hội chứng.
Tuy nhiên, chưa có phụ nữ nào có hai đột biến gen ANOS1 được báo cáo trong các tài liệu y tế. Một đặc điểm của di truyền liên kết X là người cha không thể truyền tính trạng liên kết X cho con trai của họ.
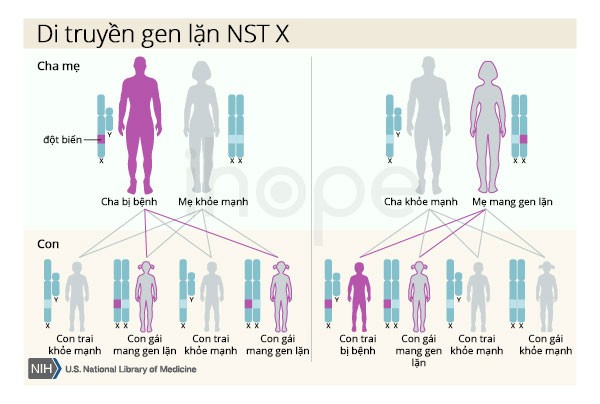
Ảnh: Cơ chế di truyền gen lặn trên nhiễm sắc thể X
Nguồn: U.S. National Library of Medicine
Thông thường, những người mắc hội chứng Kallmann do đột biến gen ANOS1 thừa hưởng đột biến từ người mẹ. Những người khác mắc hội chứng Kallmann có thể do một đột biến mới trên gen ANOS1.
Khi hội chứng Kallmann do đột biến ở các gen khác, thường có kiểu di truyền trội trên nhiễm sắc thể thường, nghĩa là một bản sao của gen bị thay đổi trong mỗi tế bào là đủ để gây bệnh. Các trường hợp khác do đột biến gen mới và xảy ra ở những người không có tiền sử mắc hội chứng này trong gia đình.

Nguồn: U.S. National Library of Medicine
Trong một số gia đình, hội chứng Kallmann có kiểu di truyền lặn trên nhiễm sắc thể thường, nghĩa là cả hai bản sao của gen trong mỗi tế bào đều có đột biến. Cha mẹ của người bệnh mang một bản sao của gen đột biến trên nhiễm sắc thể thường, nhưng họ không biểu hiện các dấu hiệu và triệu chứng của bệnh.

Nguồn: U.S. National Library of Medicine
Các tên gọi khác của Hội chứng Kallmann
- Thiểu năng sinh dục mất khứu giác
- Thiểu năng sinh dục mất khứu giác tự phát
- Suy sinh dục với mất khứu giác
- Hội chứng thiểu năng sinh dục Hypogonadotropic
- Hội chứng Kallman
Để nhận thông tin khám với Bs Nhật, vui lòng nhấn vào link: https://m.me/bsphamquangnhat